Die Vererbung der Hämophilie
Die beiden Hämophilien A und B kommen durch Mutationen in den Genen der Gerinnungsfaktoren VIII (Hämophilie A) bzw. IX (Hämophilie B) zustande. Beide Gene befinden sich auf dem X-Chromosom. Die Erkrankungen werden rezessiv vererbt, d. h. ein gesundes Allel auf einem X-Chromosom kann ein defektes Allel auf dem 2. X-Chromosom kompensieren. Das erklärt das Phänomen, dass Frauen (XX) fast immer phänotypisch gesund sind und die Erkrankung nur an männliche Nachkommen (XY) übertragen (Konduktorin). Das Vererbungsmuster ist ein wichtiges Unterscheidungskriterium zur von Willebrand-Erkrankung: Letztere wird autosomal vererbt und betrifft dadurch sowohl weibliche als auch männliche Nachkommen. Zwei Hauptmuster kennzeichnen die Vererbung der Hämophilie:
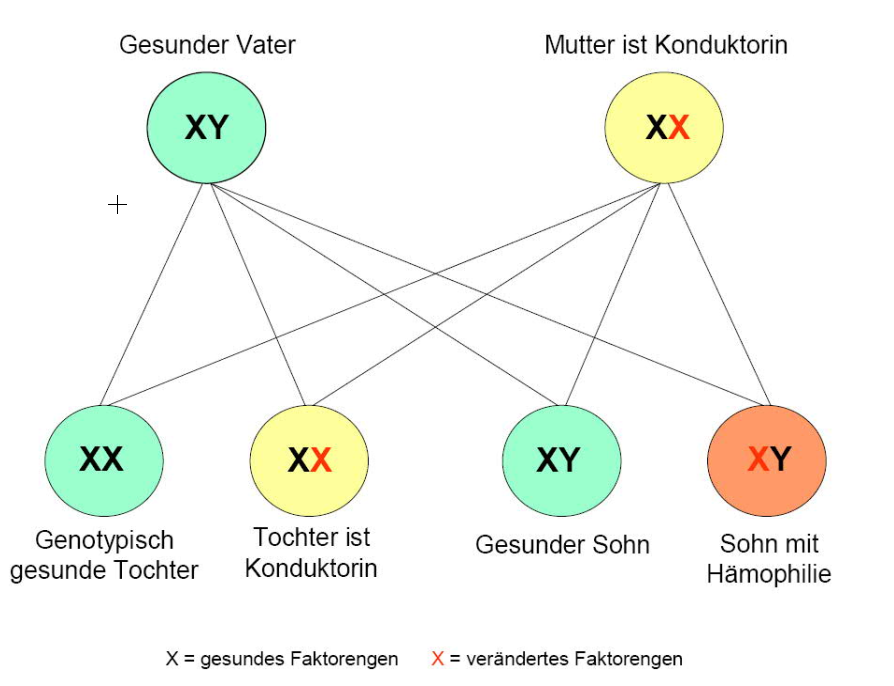
Die weiblichen Nachkommen einer Konduktorin und eines gesunden Mannes sind mit einer Wahrscheinlichkeit von 50% Konduktorin, die männlichen Nachkommen erkranken mit einer Wahrscheinlichkeit von 50% an Hämophilie.
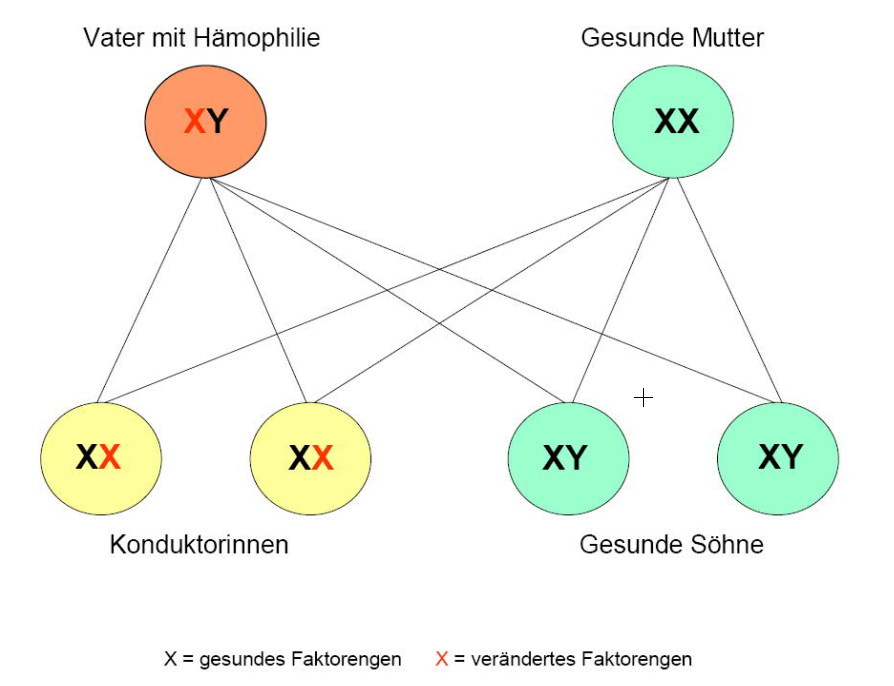
Die gemeinsamen Kinder eines Vaters mit Hämophilie und einer gesunden Mutter sind entweder gesunde Söhne oder Konduktorinnen.
In selteneren Fällen sind beide Elternteile Träger eines defekten Gens: In diesem Fall können auch weibliche Nachkommen an Hämophilie erkranken.
Häufigkeit und Schweregrade der Hämophilie
Die beiden Hämophilien A und B treten in der Bevölkerung mit unterschiedlichen Häufigkeiten auf: Die Häufigkeit der Hämophilie A bei Neugeborenen beträgt 1 : 5.000 bis 1 : 10.000 der Geburten männlichen Geschlechts, die der Hämophilie B 1 : 25.000 bis 1 : 30.000 der Geburten männlichen Geschlechts. Da das Faktor VIII-Gen etwa sechsmal größer ist als das Faktor IX-Gen, treten Spontanmutationen auch sechsmal häufiger im Faktor VIII-Gen auf.
Der Schweregrand einer Hämophilie und damit die im Blutkreislauf verbleibende Restaktivität der Faktoren VIII und IX hängt von der Schwere der Genmutationen ab und wird in Relation gesetzt zur Normalaktivität, die im Idealfall 100% beträgt :
| Schweregrad einer Hämophilie | Faktor-Restaktivität (%) |
|---|---|
| Schwere Hämophilie | < 1 |
| Mittelschwere Hämophilie | 1 – 5 |
| Leichte Hämophilie | 6 – 24 |
| Subhämophilie | 24 – 49 |
| Normalwert | 50 – 200 |
Funktionen der Faktoren VIII und IX in der Blutgerinnung
Die Faktoren VIII und IX besetzen eine wichtige Schaltstelle im hochkomplexen und streng regulierten Gerinnungssystem, an dessen Ende polymerisiertes Fibrin steht. Diese Schaltstelle bezeichnet manals Tenasekomplex ( = FX-spaltender Komplex). Faktor IX ist ein sogenanntes Proenzym und wird durch Aktivierung in das Enzym Faktor IXa überführt. Als sogenannte Serinprotease aktiviert Faktor IXa Faktor X. Die Aktivierung von Faktor X durch Faktor IXa alleine würde nur sehr langsam verlaufen.
Durch den Einfluss eines sogenannten Cofaktors wird die Reaktion um das 200.000-fache beschleunigt. Faktor VIIIa ist dieser Cofaktor. Weitere Partner für die Faktor X-Aktivierung sind die Blutplättchen oder Thrombozyten: Diese transportieren bei Aktivierung negative Ladungen an ihre Oberflächen und ermöglichen so das Andocken von aktivierten Gerinnungsfaktoren. Abbildung 3 zeigt schematisch diesen Prozess.
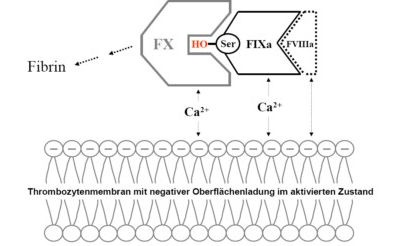
Schematische Darstellung des sogenannten Tenasekomplexes auf der Oberfläche von aktivierten Blutplättchen.
Die Behandlung der Hämophilie
Der erste historische Meilenstein in der kausalen Behandlung der Hämophilie war die Entwicklung der Bluttransfusion. Der erste Bericht der erfolgreichen Behandlung eines Hämophiliepatienten mit Blut stammt aus dem Jahr 1840, bei dem einem postoperativ blutenden Jungen etwa 300 ml („12 ounzes“) Blut einer „kräftigen jungen Frau“ transfundiert wurden. Es war schließlich R. G. Macfarlane, der in seiner 1938 vorgestellten Doktorarbeit feststellte, dass nur die Bluttransfusion – und damit die temporäre Substitution der fehlenden Komponenten – eine Behandlung von Blutungsepisoden ermöglichen konnte.
Für die effiziente Behandlung schwerer Blutungen musste die in geringer Konzentration im Blut vorkommende Komponente aufkonzentriert werden. Die Entwicklung der ersten Konzentrate verlief über (1) die Fraktionierung von Plasma durch das von Cohn entwickelte Verfahren mit graduierter alkoholischer Eiweißfällung und (2) die Entdeckung des Kryopräzipitats durch Pool und Shannon im Jahr 1965. Während die ersten Konzentrate die Behandlungsfähigkeit der Hämophilie signifikant verbesserten – erwähnt sei die Möglichkeit der Heimselbstbehandlung -, erforderten massive Komplikationen durch die Übertragung humanpathogener Viren die Entwicklung virusinaktivierter Konzentrate. Parallel dazu wurden rekombinante Faktorenkonzentrate entwickelt: Dabei wurden Faktor VIII und Faktor IX in spezialisierten Zelllinien vektoriell exprimiert. In den letzten Jahren wurden verscheiden halbwertszeitverlängerte Gerinnungsfaktoren zugelassen. Dies führte zu einer teilweise drastischen Herabsetzung der Substitutionshäufigkeit und damit zu einer Erhöhung des Behandlungskomforts. Daneben wurde eine erste disruptive Therapieform in Form eines bispezifischen Antikörpers zugelassen, die eine neue Handlungsoption bei Patienten mit Hämophilie A sowie Hämophilie-A-Patienten mit FVIII Inhibitor darstellt. Schließlich gibt es die erste Zulassung von Gentherapien bei Patienten mit Hämophilie A und B.
Die Behandlung der Hämophilie – Meilensteine aus den letzten 150 Jahren.